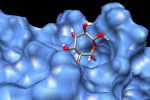
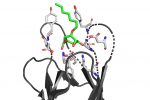
 QM/MM computed complex between ConA and a methyl septanoside.
QM/MM computed complex between ConA and a methyl septanoside.











We took our revenge on carbohydrates by attacking some cellulosic materials with axes yesterday at Pine and Iron Axe Throwing in #Hartford.

Thx @todd_hyster and @pfizer scientists for talking w/ @peczuh_research at the @ACS_CVS poster session yesterday. @swimmerkarate




@peczuh_research standing tall at today’s @ACS_CVS organic symposium. cc: @swimmerkarate

Summer Squad 2019 (minus @swimmerkarate): Jean, Bryant, Matthew, @mwpeczuh, Caleb, Yotam. We miss you CC! Come back from your @Boehringer internship already. #REU #UConnChem

